Los estados megaloblásticos se deben a la síntesis defectuosa de ADN. La síntesis de ARN continúa y, como resultado, se produce un aumento de la masa y la maduración citoplasmáticas. La circulación recibe hematíes macroovalocíticos y todas las células presentan dispoyesis, caracterizada porque la maduración citoplasmática es mayor que la nuclear, produciéndose el megaloblasto en la médula. La dispoyesis incrementa la destrucción intramedular de las células (eritropoyesis ineficaz), originándose hiperbilirrubinemia indirecta e hiperuricemia. Como están afectadas todas las líneas celulares, además de la anemia, pueden aparecer leucopenia y trombocitopenia, aunque suelen tardar en desarrollarse. Otro hallazgo característico del estado megaloblástico lo constituye la reticulocitopenia debida a la producción defectuosa de hematíes. La hipersegmentación de los neutrófilos polimorfonucleares también es un hallazgo típico, cuyo mecanismo de producción se desconoce. Además del reconocimiento morfológico de los cambios megaloblásticos, puede utilizarse la prueba de supresión de desoxiuridina para demostrar la síntesis defectuosa de ADN a nivel bioquímico.
Los mecanismos más frecuentes que causan estados megaloblásticos incluyen la utilización deficitaria o defectuosa de vitamina B12 o ácido fólico, los fármacos citotóxicos (generalmente antineoplásicos o inmunodepresores) que alteran la síntesis de ADN y una forma neoplásica autónoma rara, el síndrome de Di Guglielmo,que se considera una mielodisplasia en transformación a leucemia mieloide aguda. La identificación de la etiología y de los mecanismos fisiopatológicos de las anemias megaloblásticas resulta crucial.
Anemia por déficit de vitamina B12
La molécula de vitamina B12 consta del nucleótido 5,6-dimetilbencimidazol unido en ángulo recto con un anillo tetrapirrólico con un átomo de cobalto (el núcleo corrínico). En la naturaleza existen diversas cobalaminas (componentes de la vitamina B12) que sólo varían en el radical unido al átomo de cobalto .
La metilcobalamina (MeCbl) y la adenosilcobalamina (AdoCbl), dos coenzimas de cobalamina fisiológicas, realizan las funciones bioquímicas de la B12. La MeCbl actúa en el metabolismo del ácido nucleico y es el cofactor que interviene en la síntesis de ADN defectuoso. La AdoCbl sirve como un sistema de recogida para el catabolismo de los aminoácidos alifáticos, las membranas lipídicas y los precursores de propionato y es posible que sea el cofactor implicado en la síntesis y reparación de la mielina alterada.
La vitamina B12 está presente en la carne y los alimentos con proteínas animales. Su absorción es compleja; se lleva a cabo en el íleon terminal y requiere la presencia de factor intrínseco, una secreción de las células parietales de la mucosa gástrica, para transportar la vitamina a través de la mucosa intestinal. La vitamina B12 alimentaria se une a proteínas fijadoras (fijadoras R) de la saliva que protegen a la B12 en el medio ácido gástrico. Cuando este complejo B12 (B12-fijadoras R) se introduce en el intestino delgado, unas enzimas pancreáticas lo escinden y la vitamina B12 se une al factor intrínseco.
La vitamina B12 está presente en el plasma como MeCbl, 5´-desoxiAdoCbl e hidroxicobalamina, unida a proteínas específicas, las transcobalaminas I y II. La transcobalamina I es una forma de depósito, en tanto que la transcobalamina II es la proteína transportadora de B12 fisiológica. La concentración plasmática normal de vitamina B12 oscila entre 200 y 750 pg/ml (150-550 pmol/l), lo que sólo representa alrededor del 0,1% del contenido total del organismo, la mayoría del cual se localiza en el hígado. La excreción es principalmente biliar y, en menor grado, renal. La pérdida diaria total es de 2-5 mg; se produce cierta reutilización enterohepática.
Debido a la lenta tasa de utilización y a los considerables depósitos de vitamina B12, su deficiencia (depósitos tisulares <0,1 mg y valor sérico <150 pg/ml [110 pmol/l]) tarda en aparecer entre varios meses y años. Los depósitos hepáticos de B12 suelen bastar para satisfacer las necesidades fisiológicas durante 3-5 años en ausencia de factor intrínseco y de meses a un año en ausencia de capacidad de reabsorción enterohepática. No obstante, cuando los depósitos hepáticos son limitados y la demanda por el crecimiento es elevada, las alteraciones hematológicas y neurológicas pueden aparecer con mayor rapidez (p. ej., niños lactantes de madres vegetarianas).
Etiología y fisiopatología
La disminución de la absorción de vitamina B12 es el principal mecanismo fisiopatológico y puede deberse a varios factores. La anemia causada por deficiencia de vitamina B12 también suele denominarse anemia perniciosa. Clásicamente, el término anemia perniciosa expresa la deficiencia de B12 producida por pérdida de la secreción de factor intrínseco. La competencia por la vitamina B12 disponible y la escisión del factor intrínseco pueden ocurrir en el síndrome del asa ciega (debido al empleo bacteriano de B12) o en las infestaciones por cestodos. Las áreas de absorción ileal pueden faltar de forma congénita o destruirse por enteritis regional inflamatoria o resección quirúrgica. Causas menos frecuentes de disminución de la absorción de B12 incluyen la pancreatitis crónica, los síndromes de malabsorción, la administración de ciertos fármacos (p. ej., quelantes orales del calcio, ácido aminosalicílico, biguanidas), la ingestión inadecuada de B12 (generalmente en vegetarianos) y, en muy raras ocasiones, el aumento del metabolismo de la B12 en el hipertiroidismo de larga duración. Una causa muy habitual de deficiencia de B12 en la población anciana es la absorción inadecuada de B12 unida a alimentos en ausencia de cualquiera de los mecanismos anteriores; la vitamina B12 pura se absorbe, pero la liberación y la absorción de la B12 unida a alimentos son defectuosas.
La enfermedad sistémica combinada hace referencia a los cambios degenerativos que se producen en el sistema nervioso. Los cambios degenerativos en la sustancia blanca cerebral y en los nervios periféricos afectan tanto a los axones como a las vainas de mielina y suelen preceder a las alteraciones de las columnas posteriores y los tractos corticoespinales. Las neuronas corticales también pueden degenerar, aunque las alteraciones neuronales son menores en comparación con las que se observan en los tractos mielinizados. En ocasiones se afectan los nervios ópticos.
Síntomas y signos
La anemia generalmente se desarrolla de manera insidiosa y progresiva a medida que se agotan los depósitos hepáticos de B12. A menudo, es más intensa de lo que cabría esperar por los síntomas, porque su lenta evolución permite una adaptación fisiológica. En ocasiones se palpan esplenomegalia y hepatomegalia. Pueden estar presentes diversas manifestaciones GI, como anorexia, estreñimiento y diarrea intermitentes y dolor abdominal mal localizado. La glositis, descrita generalmente como una quemazón sobre la lengua, puede ser un síntoma temprano. Es frecuente una pérdida de peso considerable. Un signo raro es la FOD que responde con rapidez al tratamiento con B12.
Puede haber afectación neurológica incluso en ausencia de anemia. Este hecho se comprueba sobre todo en pacientes mayores de 60 años. Los nervios periféricos son los que se afectan con mayor frecuencia, seguidos de la médula espinal. Los síntomas neurológicos preceden algunas veces a las alteraciones hematológicas (e incluso ocurren en su ausencia, en especial si se ha administrado ácido fólico).
En las fases iniciales se detecta una pérdida periférica de la sensibilidad posicional y vibratoria en las extremidades, junto con debilidad leve o moderada y pérdida de reflejos. En fases posteriores aparecen espasticidad, signo de Babinski, mayor pérdida de la sensibilidad propioceptiva y vibratoria en las extremidades inferiores y ataxia. La sensibilidad táctil, algésica y térmica se alteran con menos frecuencia. Las extremidades superiores se afectan más tarde y con menos regularidad que las inferiores. Algunos pacientes también muestran irritabilidad y depresión moderada. Puede desarrollarse ceguera para los colores azul y amarillo. En los casos avanzados puede surgir paranoia (demencia megaloblástica), delirio, confusión, ataxia espástica y, en ocasiones, hipotensión postural.
Diagnóstico y datos de laboratorio
La enfermedad sistémica combinada debe diferenciarse de las lesiones medulares compresivas y de la esclerosis múltiple. El diagnóstico precoz es fundamental, ya que los defectos neurológicos se vuelven irreversibles cuando persisten durante meses o años.
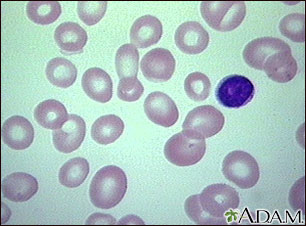
La anemia es macrocítica, con un VCM >100 fl. En la extensión se aprecia macroovalocitosis, anisocitosis y poiquilocitosis. Como es de esperar, la ADE es elevada. Es frecuente la aparición de cuerpos de Howell-Jolly (fragmentos residuales del núcleo). A menos que el paciente haya sido tratado, existe reticulocitopenia. La hipersegmentación de los granulocitos es uno de los primeros hallazgos; la neutropenia se desarrolla con posterioridad. Se observa trombocitopenia en aproximadamente la mitad de los casos graves, y las plaquetas a menudo tienen formas extrañas y tamaños desiguales. En la médula ósea se aprecian hiperplasia eritroide y cambios megaloblásticos. La bilirrubina indirecta sérica puede estar elevada como consecuencia de la eritropoyesis ineficaz y la supervivencia reducida de los hematíes defectuosos. La LDH sérica suele estar muy aumentada, lo que refleja la hematopoyesis ineficaz y el incremento de la hemólisis. La ferritina sérica está generalmente elevada (>300 ng/ml), lo cual concuerda con la existencia de hemólisis.
El método empleado con mayor frecuencia para establecer el déficit de B12 como causa de la megaloblastosis es la determinación de la vitamina B12 sérica. Si bien pueden surgir valores falsos negativos, en general, niveles inferiores a 150 pg/ml (<110 pmol/l) indican, con fiabilidad, la existencia de déficit de B12. Habitualmente, la anemia o las alteraciones neurológicas son evidentes con niveles de B12 menores de 120 pg/ml (<90 pmol/l). En circunstancias limítrofes (150-250 pg/ml [110-180 pmol/l) y cuando la sospecha clínica sugiere la existencia de una deficiencia de B12, el análisis de B12 debe complementarse con otras pruebas. La deficiencia tisular de B12 ocasiona aciduria metilmalónica (y propiónica); en consecuencia, la medición del ácido metilmalónico en suero es una prueba muy sensible para detectar el déficit de B12. Este análisis se ha convertido en la prueba de elección para el diagnóstico en caso de sospechar posibles valores falsos negativos, sobre todo en los ancianos, de los que el 5-10% tienen valores séricos de B12 normales a pesar de los indicios de deficiencia tisular. Un análisis menos habitual consiste en la determinación del contenido de transcobalamina II-B12, que identifica un equilibrio negativo de B12cuando la transcobalamina II-B12 es menor de 40 pg/ml (<30 pmol/l).
Una vez confirmada la deficiencia de B12, debe identificarse el mecanismo fisiopatológico responsable. Pueden detectarse autoanticuerpos contra las células parietales gástricas en el 80-90% de los pacientes con anemia perniciosa. Más importantes para el diagnóstico son los anticuerpos contra el factor intrínseco, que pueden hallarse en el suero de la mayoría de los pacientes. La determinación de anticuerpos antifactor intrínseco puede realizarse si el paciente no ha tomado B12 en los cinco días precedentes. La mayoría de los pacientes con anemia perniciosa presentan aclorhidria. Los análisis gástricos demuestran un pequeño volumen de secreciones gástricas (aquilia gástrica) con un pH >6,5; la aclorhidria se confirma si el pH se eleva a 6,8-7,2 tras la administración de histamina. El origen de la anemia perniciosa típica es la ausencia de secreción de factor intrínseco; éste debe determinarse en la secreción gástrica independientemente del pH, dado que puede existir una secreción discordante de ácido y de factor intrínseco.
La prueba de Schilling mide la absorción de vitamina B12 radiactiva con factor intrínseco y sin él. Es muy útil para establecer el diagnóstico en pacientes que han sido tratados y están en remisión clínica, pero en los que existen dudas respecto a la validez del diagnóstico. La prueba se realiza mediante la administración v.o. de vitamina B12 marcada radiactivamente, seguida al cabo de 1-6 h de una dosis "de refuerzo" parenteral (1.000 mg) de B12 para evitar el depósito hepático de la B12radiactiva; a continuación se determina el porcentaje de material radiactivo en la orina de 24 h (valor normal >9% de la dosis administrada). Una excreción urinaria reducida (<5% si la función renal es normal) indica una disminución de la absorción de vitamina B12. Esta prueba (Schilling I) puede repetirse (Schilling II) empleando cobalto radiactivo unido a factor intrínseco de origen porcino. La corrección de una excreción previamente reducida sugiere que la ausencia de factor intrínseco es el mecanismo fisiopatológico responsable de los valores bajos de vitamina B12. Finalmente, la incapacidad para corregir la excreción indica un mecanismo de malabsorción GI (p. ej., esprue). Puede practicarse una prueba de Schilling III tras la administración de un antibiótico oral durante 2 sem. Como la prueba produce repleción de vitamina B12, debe practicarse tras finalizar todos los estudios y ensayos terapéuticos. La prueba de Schilling no mide la absorción de B12 unida a los alimentos, por lo que no detecta los defectos de liberación de esta fracción de la vitamina en los pacientes ancianos.
Debido al aumento de la incidencia de cáncer gástrico en los pacientes con anemia perniciosa, es aconsejable practicar radiografías GI en el momento del diagnóstico. Éstas también pueden descartar otras causas de anemia megaloblástica (p. ej., divertículos o asas ciegas intestinales o los patrones característicos del intestino delgado que aparecen en el esprue). Debe realizarse una vigilancia posterior cuando los hallazgos clínicos (p. ej., síntomas, prueba de sangre oculta en heces positiva) sugieren un cambio en el estado del estómago; el papel de la endoscopia o las radiografías periódicas no está completamente definido.
Anemia por déficit de ácido fólico
Numerosos tejidos vegetales y animales contienen ácido fólico (ácido pteroilglutámico, folacina) como metil o formil poliglutamatos reducidos . En la forma tetrahidrato, los folatos actúan como coenzimas en procesos en los que existe transferencia de una unidad de carbono (p. ej., en la biosíntesis de nucleótidos purínicos y pirimidínicos), en conversiones de aminoácidos (p. ej., de histidina a ácido glutámico a través del ácido formiminoglutámico) y en la síntesis y utilización de formatos.
La absorción se lleva a cabo en el duodeno y el yeyuno proximal. En las células epiteliales, los poliglutamatos de los alimentos se reducen hasta dihidrofolatos y tetrahidrofolatos. Se unen a proteínas y se transportan como metiltetrahidrofolato. Los valores séricos oscilan entre 4 y 21 ng/ml (9-48 nmol/l) y son un fiel reflejo de la ingestión dietética. El folato eritrocitario (valores normales, 225-640 ng/ml de sangre total [510-1.450 nmol/l], corregido a un Hto del 45%) constituye un indicador más adecuado del estado tisular de folato. El folato total del organismo se aproxima a 70 mg, localizándose la tercera parte en el hígado. Alrededor del 20% del folato ingerido se excreta sin haberse absorbido, junto con 60-90 mg/d no reabsorbidos por la bilis.
Etiología y fisiopatología
La cocción prolongada destruye los folatos, que son abundantes en ciertos alimentos como vegetales de hoja verde, levaduras, hígado y setas. En ausencia de ingestión, los depósitos hepáticos sólo proporcionan suministro durante 2-4 meses. Es habitual la ingestión dietética limitada de ácido fólico. El alcohol interfiere en su metabolismo intermediario, absorción intestinal y circulación enterohepática. Por esta razón, las personas que siguen una dieta carencial (p. ej., "té y tostadas", alcohólicos crónicos) son propensas a desarrollar una anemia macrocítica por déficit de folato, al igual que aquellos que padecen una hepatopatía crónica. Dado que el feto obtiene el ácido fólico por suministro materno, las mujeres gestantes son susceptibles de desarrollar una anemia megaloblástica.
La malabsorción intestinal es otra causa frecuente de deficiencia de folato. En el esprue tropical, la malabsorción es secundaria a la atrofia de la mucosa intestinal resultante de la carencia de ácido fólico, por lo que incluso dosis mínimas suelen corregir la anemia y la esteatorrea. El déficit de folato puede desarrollarse en pacientes tratados con anticonvulsivantes o anticonceptivos orales durante períodos prolongados debido a la disminución de la absorción, así como en individuos en tratamiento con antimetabolitos (metotrexato) y fármacos antimicrobianos (p. ej., trimetoprim/sulfametoxazol) que alteran el metabolismo del folato. Finalmente, el aumento de la demanda de folato se produce en la gestación y la lactancia, en las anemias hemolíticas crónicas (sobre todo congénitas), en la psoriasis y en la diálisis crónica.
Diagnóstico
Las manifestaciones clínicas principales son las propias de la anemia. La deficiencia de folato es indistinguible de la de vitamina B12 por los hallazgos en sangre periférica y en médula ósea, pero no se observan las lesiones neurológicas propias del déficit de B12. El folato es fundamental en la formación del sistema nervioso durante los períodos fetal y neonatal. Cuando no se ingiere folato durante el embarazo pueden surgir defectos del tubo neural con alteraciones neurológicas graves. Otros síntomas neurológicos poco frecuentes (síndrome de piernas inquietas del embarazo) también se han relacionado con la deficiencia de folato. La principal prueba de laboratorio para diferenciar esta deficiencia de otras formas clínicas de anemia megaloblástica consiste en medir la depleción de folato. Concentraciones séricas de ácido fólico <4 ng/ml (<9 nmol/l) sugieren deficiencia; el hallazgo de valores bajos de folato eritrocitario (valores normales, 225-600 ng/ml [510-1.360 nmol/l]) confirma el déficit tisular. (El intervalo de normalidad depende del método de laboratorio empleado.) Ambas determinaciones presentan resultados falsos positivos y falsos negativos. Como consecuencia, la medición de homocisteína sérica proporciona el mejor dato sugestivo de deficiencia tisular. No obstante, dado que la B12 utiliza la misma vía, deben determinarse el ácido metilmalónico y la homocisteína. Un valor normal de ácido metilmalónico con una cifra elevada de homocisteína confirma el diagnóstico de déficit de folato.
dispoyesis: anomalía en la maduración de todos los tipos celulares. En la anemia macrocítica megaloblástica el VCM es mayor de 110 fentolitros y las células presentan una mayor maduración citoplasmática que nuclear (dispoyesis).